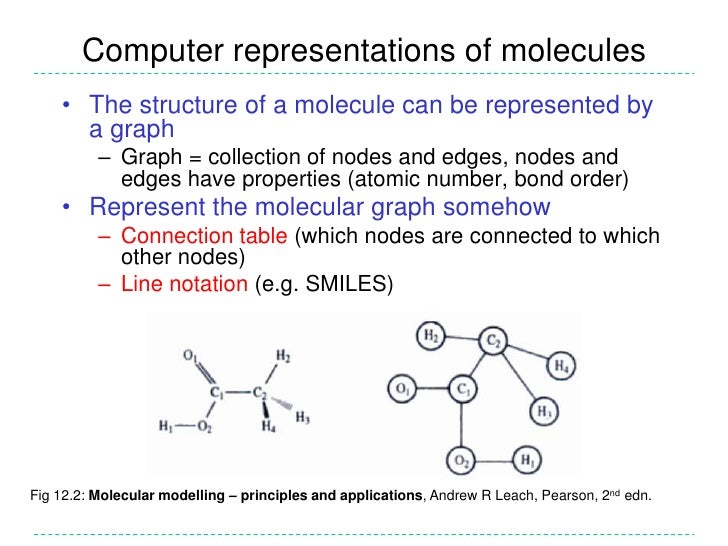
Molecular Descriptors For Cheminformatics Pdf To Jpg
In the post-genomic era, the role informatics technologies plays in chemical and pharmaceutical research has become increasingly important in data management and lead-compound identification. In Chemoinformatics: Concepts, Methods, and Tools for Drug Discovery, well-recognized pioneers and investigators from diverse professional environments survey the key concepts in the field, describe cutting-edge methods, and provide exemplary pharmaceutical applications. The authors explain the theory behind the crucial concepts of molecular similarity and diversity, describe the challenging efforts to use chemoinformatics approaches to virtual and high-throughput screening, and illuminate the latest developments in multidimensional QSAR analysis. Other topics of interest include the use of partitioning algorithms and classification methods for analyzing large compound databases, screening sets, and virtual screening for active molecules; different approaches to target class-specific library design; the generation of novel classes of molecular descriptors; and Web-based tools for chemical database access and management. Also presented are different methods for describing molecular chirality and conformational parameters and for predicting the drug-like character and basic ADME properties of compounds based on modeling their putative interactions with cytochrome P450 isoforms.
Most of the example on this page are related to the CDK package, an opensource framework for cheminformatics. Conformer data 3D similarity Fragmenting a molecule SMILES to SDF Molecular Descriptor GUI Surface areas in the CDK Geometric transformations Reading molecules from disk Writing molecules. Dec 13, 2011. The CDK-Taverna 2.0 plug-in provides 192 workers for input and output (I/O) of various chemical file and line notation formats, substructure filtering, aromaticity detection, atom typing, reaction enumeration, molecular descriptor calculation and data analysis. Parallel computing with multi-core processors.
State-of-the-art and user-friendly, Chemoinformatics: Concepts, Methods, and Tools for Drug Discovery illuminates the conceptual and methodological diversity of this rapidly evolving field and offers instructive examples of cutting-edge applications in the drug discovery process.

The ANTARES dataset is a large collection of known and verified experimental bioconcentration factor data, involving 851 highly heterogeneous compounds from which 159 are pesticides. The BCF ANTARES data were used to derive a conformation-independent QSPR model. A large set of 27,017 molecular descriptors was explored, with the main intention of capturing the most relevant structural characteristics affecting the studied property. The structural descriptors were derived with different freeware tools, such as PaDEL, Epi Suite, CORAL, Mold 2, RECON, and QuBiLs-MAS, and so it was interesting to find out the way that the different descriptor tools complemented each other in order to improve the statistical quality of the established QSPR. The best multivariable linear regression models were found with the Replacement Method variable sub-set selection technique. The proposed QSPR model improves previous reported models of the bioconcentration factor in the present dataset. The Elder Scrolls V Skyrim Crack Razor1911 Download more.